Introduction
Angelman Syndrome is a rare neurological disorder that affects roughly 1 in every 15,000 people. Classic symptoms of Angelman Syndrome include developmental delay, speech and balance impairments, excessive excitability, hand flapping, and trouble with sleep. However, only some cases of Angelman Syndrome display additional symptoms, such as epilepsy. It is known that Angelman Syndrome patients with epilepsy have a specific mutation in the gene, SNRPN, but it is unknown what role this mutation plays in disease.
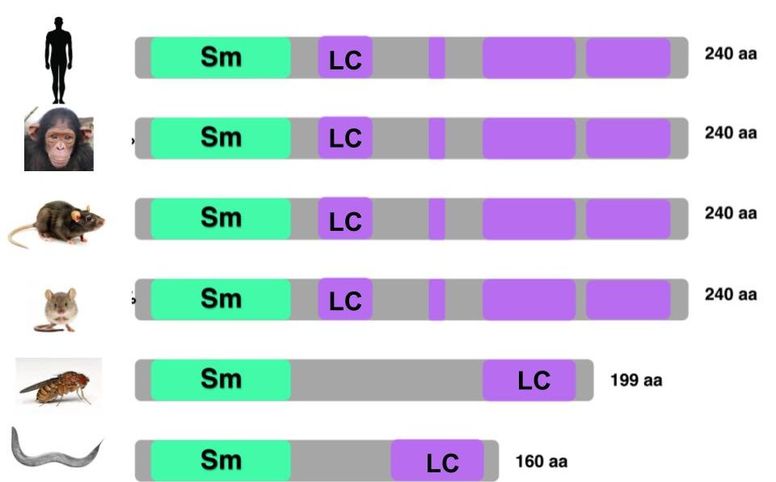
SNRPN is a protein conserved across a diverse array of species, and consists of an Sm domain, which is implicated in RNA binding, and multiple Low Complexity (LC) domains of unknown function (Figure 1).
SNRPN is a protein conserved across a diverse array of species, and consists of an Sm domain, which is implicated in RNA binding, and multiple Low Complexity (LC) domains of unknown function (Figure 1).
|
|
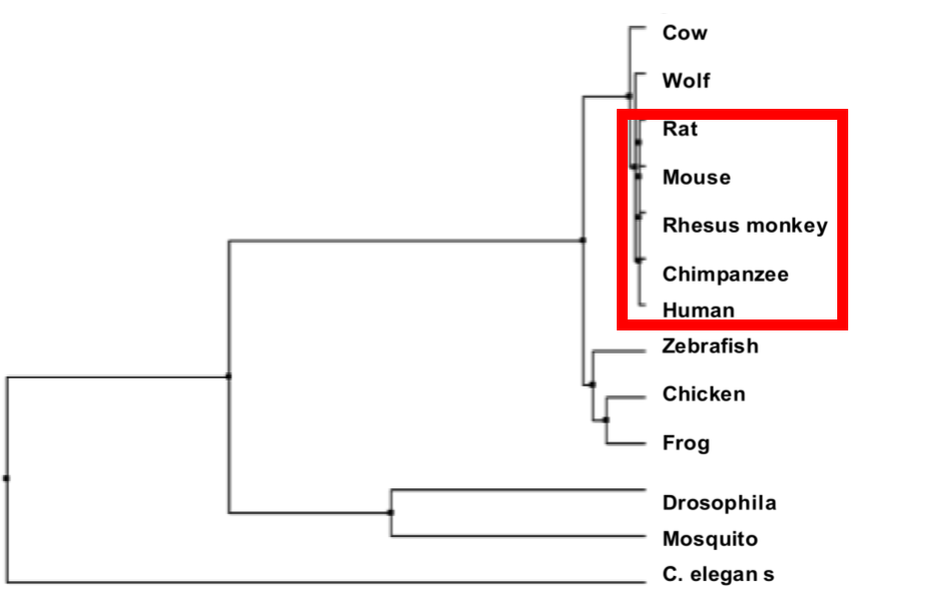
Figure 1. Left: SNRPN homologs in different species. Right: Phyolgenetic relationships of SNRPN homologs. Red box indicates species with 100% similarity between homolog sequences.
Through the Gene Ontology database, I found that SNRPN primarily functions as an RNA processing enzyme that is present at high levels in neurons. Its molecular function is RNA binding through the Sm domain, its biological process is RNA processing via the spliceosome, and its cellular component is the nucleoplasm.
Figure 2. The molecular function of SNRPN is RNA binding.
|
|
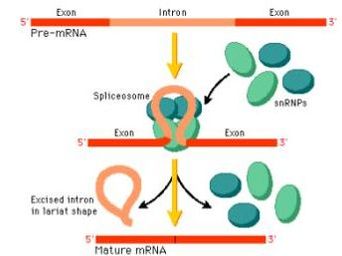
As a component of the spliceosome, SNRPN interacts with an extensive number of proteins (Figure 5). This includes not only other direct components of the spliceosome, but also scaffolding proteins and regulatory proteins. Because of the many components involved in RNA processing and spliceosome assembly, it can be difficult to determine what specific proteins are critical for proper function. With this in mind, SNRPN function in neurons can be analyzed in the context of epilepsy in cases of Angelman Syndrome.
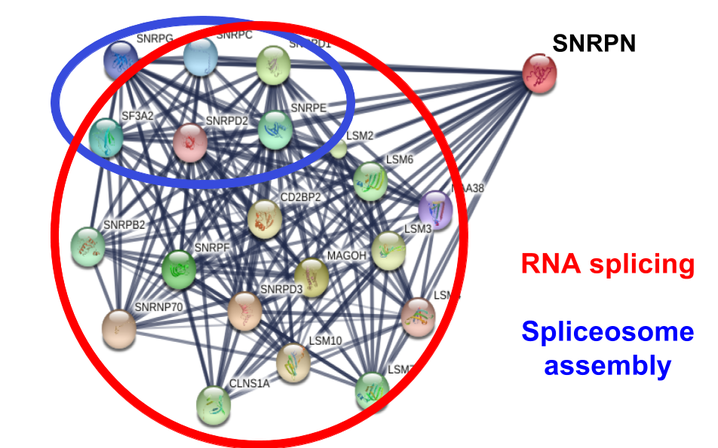
Figure 5. Human SNRPN and its protein interaction network.
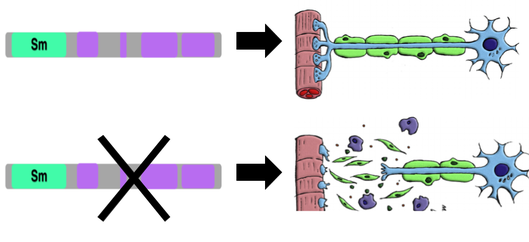
It is known that improper splicing in neurons can lead to neurodegeneration, which may result in downstream epilepsy. Although epileptic Angelman Syndrome patients have specific SNRPN mutations, it is unknown what role SNRPN plays in proper neuronal function.
My goal is to determine what is the role of RNA processing in neuronal function.
My hypothesis is that SNRPN is critical for RNA processing, and SNRPN dysfunction leads to epilepsy.
My long-term goal is to determine if defects in SNRPN RNA processing is responsible for other phenotypic effects in patients with Angelman Syndrome.
To investigate this goal and hypothesis, I have proposed a series of experiments, or Specific Aims, that will address different components of the role of SNRPN in RNA processing, and detect connections to epilepsy. All of these experiments will be conducted in mice because not only is the mouse SNRPN homolog 100% identical to human SNRPN, but also the SNRPN mutant phenotype in mice is epilepsy. Furthermore, epilepsy can be easily quantified and detected in mice using an EEG test (Figure 6).
My goal is to determine what is the role of RNA processing in neuronal function.
My hypothesis is that SNRPN is critical for RNA processing, and SNRPN dysfunction leads to epilepsy.
My long-term goal is to determine if defects in SNRPN RNA processing is responsible for other phenotypic effects in patients with Angelman Syndrome.
To investigate this goal and hypothesis, I have proposed a series of experiments, or Specific Aims, that will address different components of the role of SNRPN in RNA processing, and detect connections to epilepsy. All of these experiments will be conducted in mice because not only is the mouse SNRPN homolog 100% identical to human SNRPN, but also the SNRPN mutant phenotype in mice is epilepsy. Furthermore, epilepsy can be easily quantified and detected in mice using an EEG test (Figure 6).
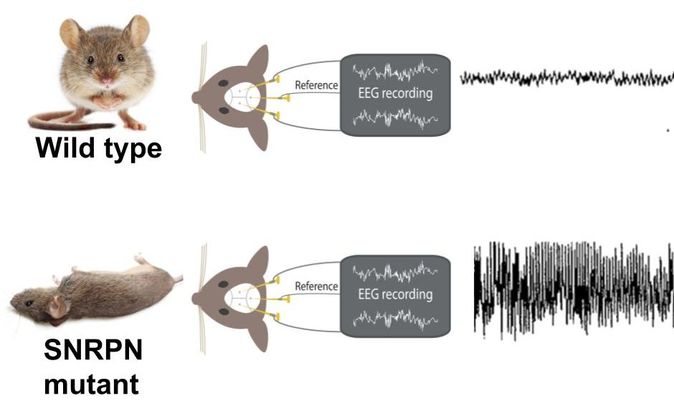
Figure 6. EEG epilepsy test comparisons between wildtype/non-epileptic mice (top) and SNRPN mutant/epileptic mice (bottom>
Specific Aims
|
Aim 1: Determine which SNRPN domains are necessary for proper RNA processing in neurons.
Through SMART database, it was found that all SNRPN homologs contain a conserved Sm domain as well as Low Complexity (LC) domains. Because these domains are conserved across diverse species, it is likely that these domains are important to SNRPN function. Aim 1 will determine which of these SNRPN domains are crucial for proper RNA processing in neurons, where neuronal function will be assessed through an epilepsy test. To accomplish this, I will create a mouse model with a single domain knockout that is generated through CRISPR/Cas9 techniques. As a result, I will have engineered knockout mutants for the Sm domain and each type of LC domain. To test for epilepsy, a determinant of neuronal function, I will conduct an EEG test on each type of knockout mutant, and compare the results to an EEG of a wildtype mouse. I hypothesize that the Sm domain of SNRPN is required for RNA binding and the LC domains will be required for associating with the spliceosome complex and other regulatory proteins, therefore inhibiting the function of either of these domains will prevent proper neuronal RNA processing from occurring, leading to epilepsy (Figure 7). Aim 2: Determine if domain mutants of SNRPN lead to differential expression of neuronal genes involved in RNA processing. It can be beneficial to compare gene expression between samples by sequencing the RNA transcripts that are present. By determining what transcripts are present in a sample, it can be determined what genes are required under certain conditions. This method can also be used to compare healthy and diseased samples to identify the difference in gene expression between the two. These differences may indicate which genes/gene products contribute to disease. I will apply this idea in the context of SNRPN. Similar to Aim 1, I will generate specific SNRPN domain mutants in mice using CRISPR/Cas9, and perform RNA-seq on the neuronal tissue of each mutant as well as on the wildtype neuronal tissue and analyze gene expression (Figure 8). Mutants with differential expression will be examined for epilepsy using an EEG test to determine if alternate gene expression disrupts neuronal function. Following this, I will then use PANTHER database to determine what gene ontology groups are deferentially expressed in epileptic mice. If differential expression is evident in mutants compared to wildtype, it may indicate that SNRPN mutations are responsible. Furthermore, if domain mutants are enriched for alternate transcripts are also epileptic, the gene ontology groups with differential expression may also be responsible for neuron function (Figure 9). I hypothesize that Sm domain mutants will not be able to bind RNA and LC domain mutants will not be able to properly associate with spliceosome proteins, therefore these mutants will not be able to process neuronal transcripts and result in epilepsy. In addition, epileptic mice will alternate expression for genes involved in splicing, locomotion, and stimulus response compared to wildtype. Aim 3: Identify novel protein interactors of SNRPN that are critical for RNA processing in motor neurons. Wildtype SNRPN interacts with an extensive network of proteins, including components of the spliceosome and various regulatory and scaffolding proteins. This protein network is typically essential to the proper function of the protein. By investigating what proteins interact with wildtype SNRPN compared to mutant SNRPN, it may indicate what proteins are required for proper RNA processing and neuronal function. To determine this, I will conduct AP-MS to isolate and identify SNRPN protein interactors through co-immunoprecipitation in neurons of wild type mice and domain mutants generated similarly to Aim 1 and Aim 2 through CRISPR/Cas9 techniques (Figure 10). I will compare the protein interactors among all samples and isolate the proteins that can interact with wild type mice but not the domain mutants. I will then identify the interactors by mass spectrometry (Figure 11). I will subsequently use CRISPR to knockout these novel interactors and perform an EEG epilepsy test to determine if the protein is required for neuronal function (Figure 12). I hypothesize that SNRPN Low Complexity (LC) domains will be involved in interactions with spliceosome regulatory and complex proteins required for neuronal RNA processing. CRISPR knockouts of these proteins will result in epileptic mice. Sm domain mutants will still be able to associate with interactors comparable to wildtype because the Sm domain will be required for RNA binding rather than protein-protein interactions. |
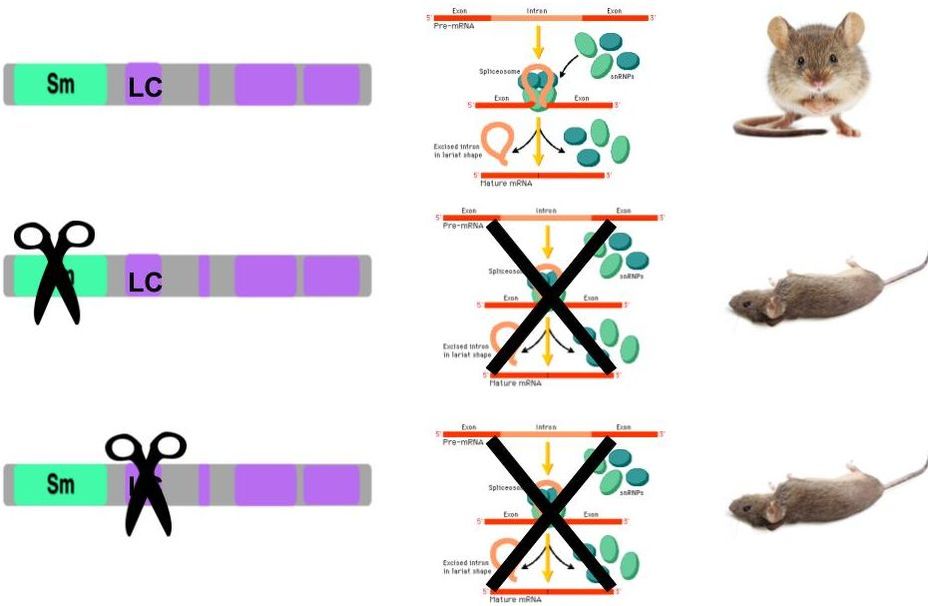
Figure 7. Domain knockouts generated by CRISPR/Cas9 are hypothesized to disrupt RNA processing in neurons, resulting in epileptic mice.
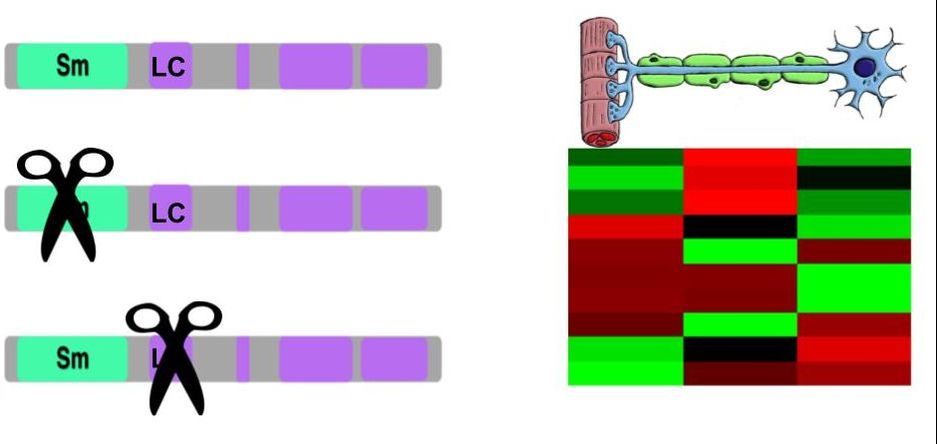
Figure 8. Aim 2 experimental approach. CRISPR/Cas9 generated domain knockouts (left) will undergo RNA-seq on their neuronal tissue samples (right).
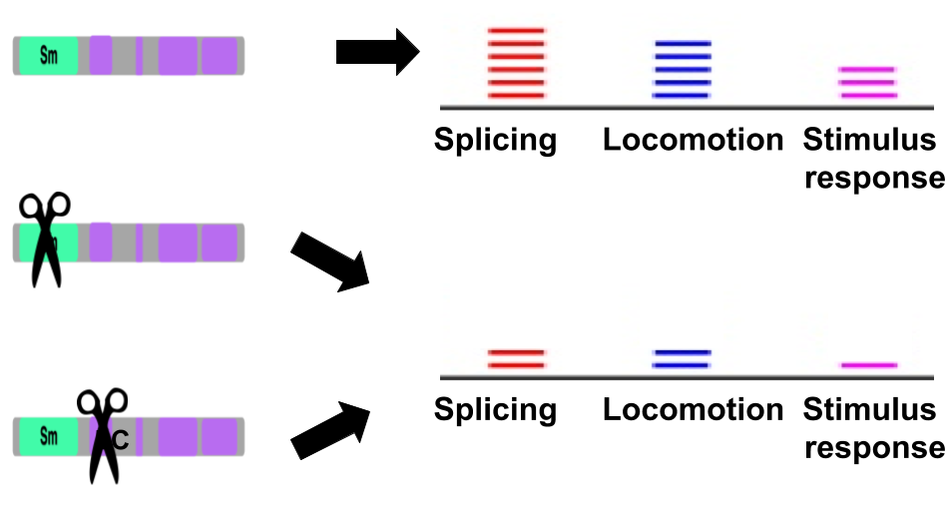
Figure 9. Domain mutants will be enriched for alternate gene ontology groups compared to wildtype.
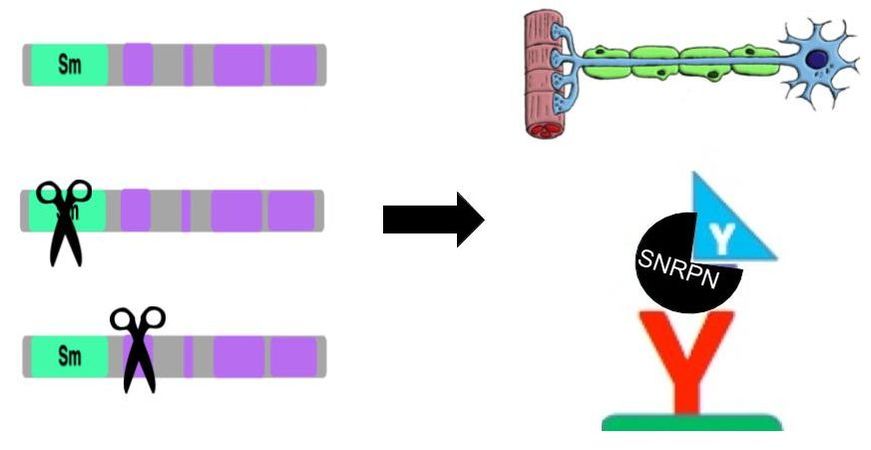
Figure 10. Protein interactors of SNRPN will be assessed in neuronal tissues of wildtype mice as well as domain mutant mice.
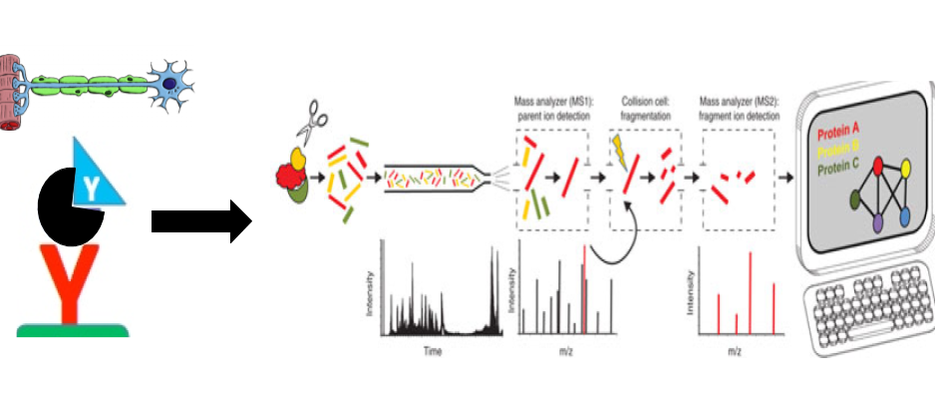
Figure 11. SNRPN interactors will be identified via affinity purification-mass spectrometry (AP-MS).
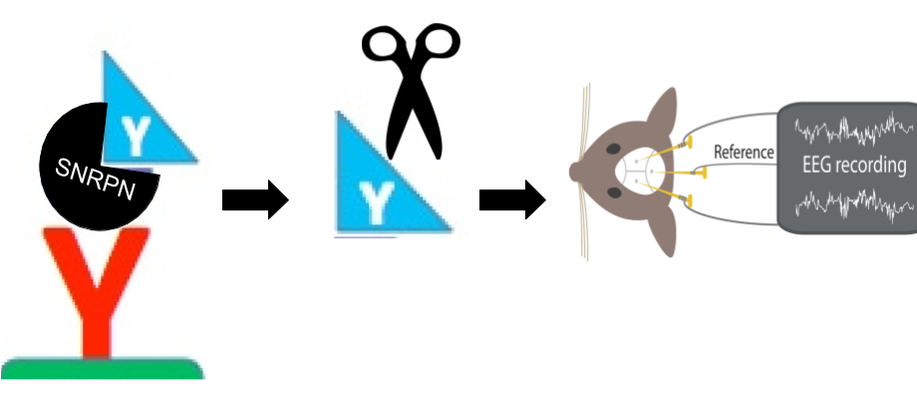
Figure 12. Isolated interactors will be knocked out via CRISPR/Cas9 techniques and assessed for neuronal dysfunction through an EEG test.
|
Future Directions
Going forward, I would design an experiment to determine if SNRPN mutants can be rescues. To asses this, I would perform a chemical screen on different SNRPN domain mutants to identify any potential small molecules that ameliorate epileptic phenotypes in mutant mice (Figure 13).
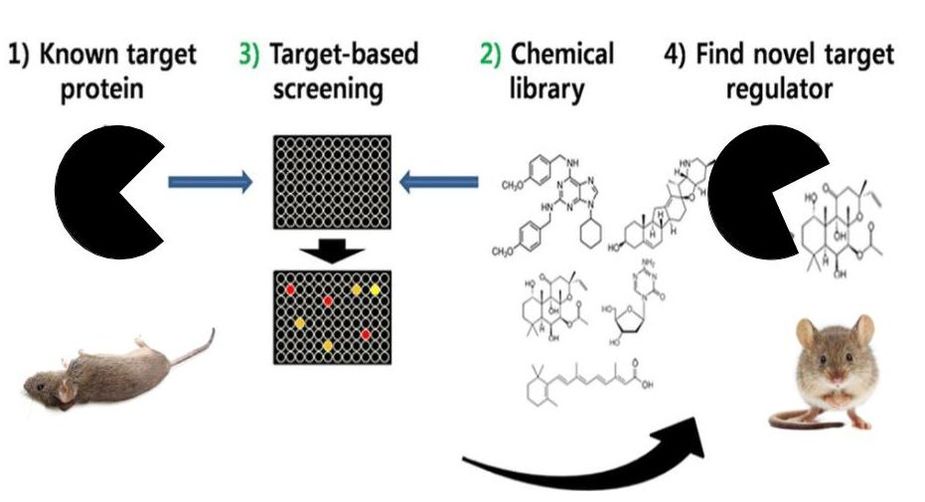
Figure 13. Chemical screen identifying small molecules with therapeutic potential.
Talk Downloads
| fischer_finalpresentation_27april2017.pptx.pdf |
| fischer_presentationdraft2_4-4-17.pptx |
| fischerjulie_21feb2017_draftpresentation__1_.pdf |
References
[1] Angelman Syndrome: Genetics Home Reference
<https://ghr.nlm.nih.gov/condition/angelman-syndrome>
[2] Li, H., Zhao, P., Xu, Q., Shan, S., Hu, C., Qiu, Z., & Xu, X. (2016). The autism-related gene SNRPN regulates cortical and spine development via controlling nuclear receptor Nr4a1. Scientific Reports, 6, 29878. doi:10.1038/srep29878
<https://www.ncbi.nlm.nih.gov/pubmed/1533223>
[3] Neuropathology: An illustrated interactive course for medical students and residents <http://neuropathology-web.org/chapter9/chapter9hAtaxia.html>
[4] Gallo, J. . (2005). The role of RNA and RNA processing in Neurodegeneration. Journal of Neuroscience, 25(45), 10372–10375. doi:10.1523/jneurosci.3453-05.2005
< http://www.jneurosci.org/content/25/45/10372 >
[5] Battaglia, A., Gurrieri, F., Bertini, E., Bellacosa, A., Pomponi, M. G., Paravatou-Petsotas, M., . . . Neri, G. (1997). The inv dup(15) syndrome: A clinically recognizable syndrome with altered behavior, mental retardation, and epilepsy. Neurology, 48(4), 1081-1086. doi:10.1212/wnl.48.4.1081
<https://www.ncbi.nlm.nih.gov/pubmed/9109904>
[6] Johnstone, K. A. (2005). A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Human Molecular Genetics, 15(3), 393-404. doi:10.1093/hmg/ddi456
<https://www.ncbi.nlm.nih.gov/pubmed/16368707?dopt=Abstract>
<https://ghr.nlm.nih.gov/condition/angelman-syndrome>
[2] Li, H., Zhao, P., Xu, Q., Shan, S., Hu, C., Qiu, Z., & Xu, X. (2016). The autism-related gene SNRPN regulates cortical and spine development via controlling nuclear receptor Nr4a1. Scientific Reports, 6, 29878. doi:10.1038/srep29878
<https://www.ncbi.nlm.nih.gov/pubmed/1533223>
[3] Neuropathology: An illustrated interactive course for medical students and residents <http://neuropathology-web.org/chapter9/chapter9hAtaxia.html>
[4] Gallo, J. . (2005). The role of RNA and RNA processing in Neurodegeneration. Journal of Neuroscience, 25(45), 10372–10375. doi:10.1523/jneurosci.3453-05.2005
< http://www.jneurosci.org/content/25/45/10372 >
[5] Battaglia, A., Gurrieri, F., Bertini, E., Bellacosa, A., Pomponi, M. G., Paravatou-Petsotas, M., . . . Neri, G. (1997). The inv dup(15) syndrome: A clinically recognizable syndrome with altered behavior, mental retardation, and epilepsy. Neurology, 48(4), 1081-1086. doi:10.1212/wnl.48.4.1081
<https://www.ncbi.nlm.nih.gov/pubmed/9109904>
[6] Johnstone, K. A. (2005). A human imprinting centre demonstrates conserved acquisition but diverged maintenance of imprinting in a mouse model for Angelman syndrome imprinting defects. Human Molecular Genetics, 15(3), 393-404. doi:10.1093/hmg/ddi456
<https://www.ncbi.nlm.nih.gov/pubmed/16368707?dopt=Abstract>
