This web page was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
What is phylogeny?
Phylogeny is study of the evolutionary relationship of organisms [1]. Phylogeny involves establishing relationship between DNA or protein sequences and constructing a phylogenetic tree based on these relationships [2]. This page displays different phylogenetic relationship analyses between SNRPN homologs, which are also explained below.
How are phylogenetic relationships determined?
To construct phylogenetic trees, relationships between homologs of different species must be determined. To accomplish this, FASTA formats of the homolog sequences are compared. For SNRPN and its homologs see the FASTA fomats below:
Once FASTA formats are determined, a program called Clustal Omega can be used to align the sequences to identify similarities and differences between the homologs. The Clustal Omega sequence alignment between SNRPN and its homologs can be seen below:





Figure 1. SNRPN homolog sequence alignments via Clustal Omega [3].
|
|
Different letters in the alignment correspond to different amino acids within the protein. Different colors are used so that sequence differences can be easily identified. Regions of the protein with a dominant color/letter among the majority of homologs indicate particular sequences that are heavily conserved. Conservation may indicate that the given region is important for proper biological function.
Using these sequence alignments, different types of phylogenetic trees can be constructed:
Percent Identity vs. BLOSUM Matrix
Phylogenetic trees can be constructed using two main approaches: Percent Identity and BLOSUM Matrix.
Percent Identity: the percentage of similarity between two gene or protein sequences [4].
BLOSUM Matrix: a scoring method used to examine the similarity between two protein sequences, denoting different scores when comparing amino acids between proteins. The scoring is based on similarity are designated based on whether the matches are likely to occur by chance [4].
Percent Identity: the percentage of similarity between two gene or protein sequences [4].
BLOSUM Matrix: a scoring method used to examine the similarity between two protein sequences, denoting different scores when comparing amino acids between proteins. The scoring is based on similarity are designated based on whether the matches are likely to occur by chance [4].
Neighbor-Joining vs. Average Distance
Neighbor-Joining and Average Distance methods are used to construct the actual phylogenetic tree based on the relationships established through the Percent Identity or BLOSUM Matrix analyses.
Neighbor-Joining: uses sequence analyses to determine how closely related homologs are, and then constructs a tree with different branch lengths, based on the magnitude of sequence changes that occurred from the time the two species diverged [4].
Average Distance: uses sequence analyses to determine how closely related homologs are, and then constructs a tree with equal branch lengths, assuming sequence changes between diverging species are equal [4].
Neighbor-Joining: uses sequence analyses to determine how closely related homologs are, and then constructs a tree with different branch lengths, based on the magnitude of sequence changes that occurred from the time the two species diverged [4].
Average Distance: uses sequence analyses to determine how closely related homologs are, and then constructs a tree with equal branch lengths, assuming sequence changes between diverging species are equal [4].
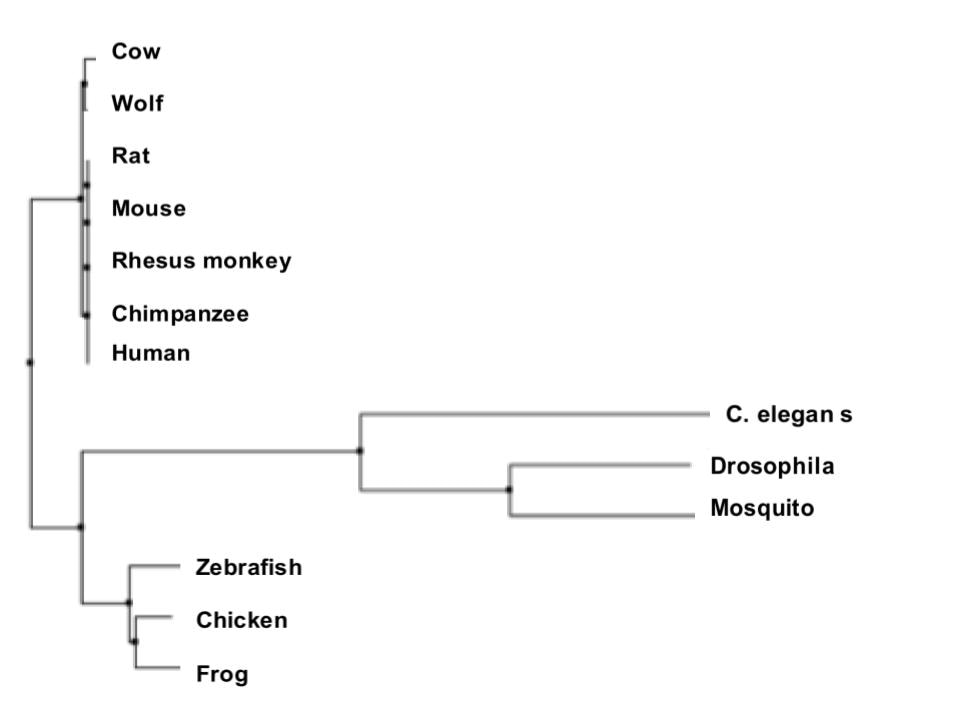
Above is an example of a phylogenetic tree created by the Neighbor-Joining method using Percent Identity [5].
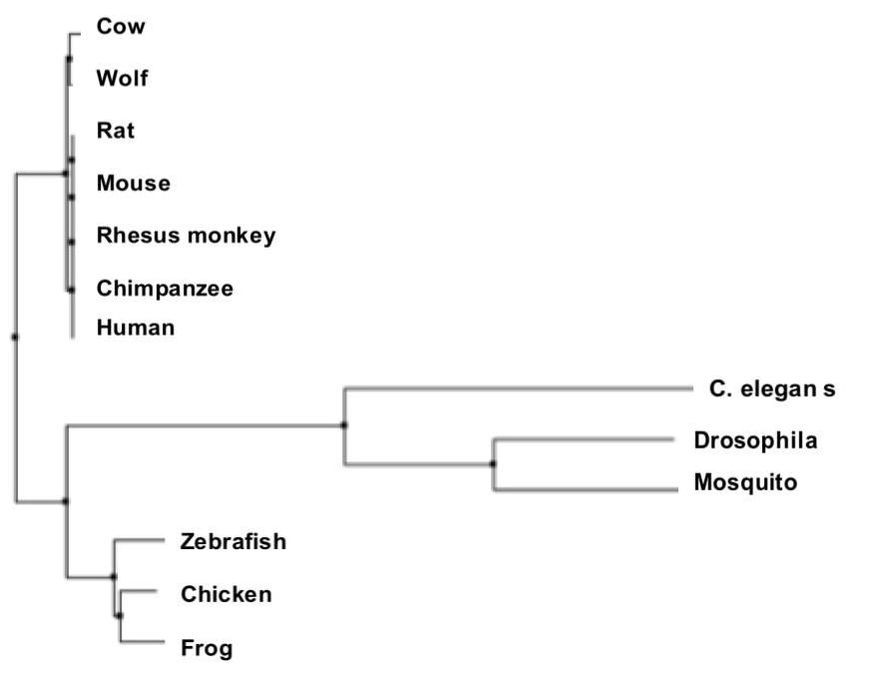
Above is an example of a phylogenetic tree created by the Average Distance method using Percent Identity [5].
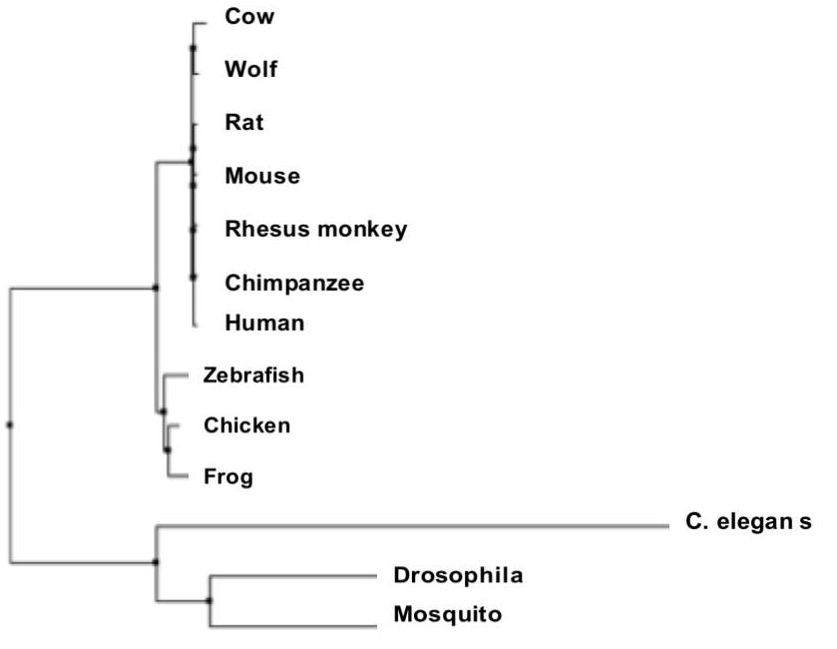
Above is an example of a phylogenetic tree created by the Neighbor-Joining method using a BLOSUM Matrix [5].
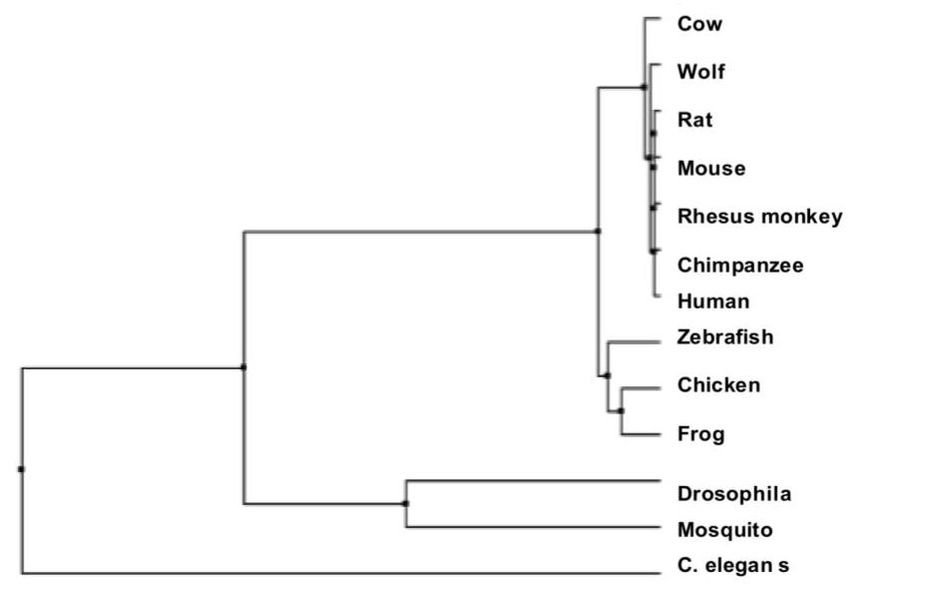
Above is an example of a phylogenetic tree created by the Average Distance method using a BLOSUM Matrix [5].
Discussion
SNRPN is highly conserved, functioning across a diverse group of species. Although the human homolog is more closely related to the SNRPN homolog of other vertebrates, SNRPN still has a phylogenetic relationship with invertebrates as well. Because of its extensive conservation, this indicates that SNRPN is important for proper biological function in many domains of life.
References
[1] <http://evolution.berkeley.edu/evolibrary/article/phylogenetics_02>
[2] <http://tolweb.org/tree/learn/concepts/whatisphylogeny.html>
[3] <http://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20170510-194848-0159-35586115-oy&tool=clustalo&showColors=true>
[4] <http://doebleygen564s14.weebly.com/how-to-make-a-phylogenetic-tree.html>
[5] <http://www.ebi.ac.uk/Tools/msa/clustalo/>
[2] <http://tolweb.org/tree/learn/concepts/whatisphylogeny.html>
[3] <http://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20170510-194848-0159-35586115-oy&tool=clustalo&showColors=true>
[4] <http://doebleygen564s14.weebly.com/how-to-make-a-phylogenetic-tree.html>
[5] <http://www.ebi.ac.uk/Tools/msa/clustalo/>
